


clinical care
Identify Sickle Cell Disease
You can play a role in diagnosing sickle cell disease and prevent vision loss by recognizing its ocular manifestations.
ALEXANDRA M. ESPEJO O.D., F.A.A.O.,
DIANA L. SHECHTMAN O.D., F.A.A.O., AND
SHERROL A. REYNOLDS O.D., F.A.A.O.,
Ft. Lauderdale, Fla.
More than 70,000 Americans have sickle cell disease, and roughly 1,000 U.S. babies are born with the disease each year, according to the Sickle Cell Disease Association of America, Inc.1
Because sickle cell disease results in ocular tissue damage, you're in an excellent position to not only prevent possible resulting vision loss, but also identify the presence of the disease in an undiagnosed patient. These actions, in turn, reinforce your value as a critical member of the healthcare team.
Here, we discuss the disease itself, its ocular manifestations and how you should manage these patients.
Sickle cell disease overview
Sickle cell disease is an inherited disorder caused by a mutation in the hemoglobin gene. Normal hemoglobin is oval and pliable, allowing it to easily travel through the microvasculature portion of the circulatory system. In sickle cell disease, the abnormal hemoglobin is "sickle" or crescent-like. As a result, its ability to carry oxygen is limited; it may also disturb normal blood flow, and it increases blood viscosity.
Due to its rigid shape, sickle-shaped hemoglobin often results in chronic hemolysis and vaso-occlusive events, such as a retinal vein occlusion. The hemolysis and vaso-occlusive events, primarily occurring under an ischemic setting, set forth a vicious cycle resulting in systemic and ocular tissue damage.2
In the United States, sickle cell disease has a predilection for black and Mediterranean individuals.3 Sickle cell disease has four main variants based on genotype.4 They are: sickle cell trait (AS), sickle cell thalassemia disease (S-Thal), sickle cell C disease (SC) and sickle cell anemia "hemoglobin S" (SS) (see "Sickle Cell Anemia Patient," below).
Sickle cell anemia "hemoglobin S" (SS) is more commonly associated with systemic complications, such as spleen enlargement, among other organ damage. On the other hand, sickle cell thalassemia (S-thal) and sickle cell C disease (SC) are more frequently associated with ocular manifestations.3-4
Although rare, ocular complications can also present in individuals who have the sickle cell trait (see "Sickle Cell Disease Suspect," below).
| Sickle Cell Anemia Patient |
|---|
| A 41-year-old black woman presented for a comprehensive eye exam without any ocular or visual complaints. Her ocular history was significant for a prosthetic right eye as a result of herpes zoster ophthalmicus (HZO) retinopathy. Her medical history revealed a diagnosis of sickle cell anemia. Exam Findings The patient's best-corrected visual acuity (BCVA) was 20/20 O.S. Slit lamp evaluation revealed no pathology. Dilated fundus evaluation O.S., however, revealed a macular pseudohole secondary to a tractional retinal detachment near the macula. (See figure 3). In addition, during our evaluation of the peripheral retina, we noted fibrovascular tissue with associated arteriovenous anastomoses (see figure 1). 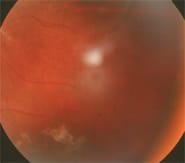
Fig 1: retinal vascular anastomoses. Diagnosis As a result of these findings, we diagnosed the patient as having proliferative sickle cell retinopathy (SCR). Management We referred the patient to her primary-care physician for continuous management of her Sickle cell anemia and to a retina specialist for further evaluation. Since the patient's vision was unaffected and she was monocular, the retinal specialist recommended a conservative approach, which consisted of continuous follow-up visits every three months. |
Ocular manifestations
The ocular manifestations of sickle cell disease involve both the anterior and posterior segment.
Anterior segment findings: comma-shaped capillaries (a classic finding denoting the accumulation of sickle cells within the capillaries), lid edema, conjunctival injection and anterior ischemia associated with iris atrophy and iris neovascularization.4-7
Posterior segment "involvement" indicates a greater propensity than anterior segment involvement toward vision loss associated with sickle cell retinopathy (SCR).8
SCR has two classifications: non-proliferative and proliferative. Retinal hemorrhages (salmon patches) that result from ischemic vasculopathy are characterized as non-proliferative retinopathy. The salmon color represents the evolution of the original bright intraretinal hemorrhage. As these hemorrhages resolve, they may gain access to the subretinal space, disrupting the retinal pigment epithelium (RPE) and, therefore, causing the development of a black sunburst lesion. The black sunburst lesion results from RPE migration forward into the intra-retinal space.
Additional non-proliferative posterior segment involvement: vascular alterations associated with venous tortuosity, angioid streaks and arterial sclerosis.5-6,8
Clinicians commonly observe arterial sclerosis adjacent to the area of capillary dropout.
Further vascular signs: occlusive retinal disease, such as retinal vein and artery occlusions, represent direct obstruction by the "sickle" hemoglobin. Retinal vein occlusions and artery occlusions are often associated with decreased visual acuity. In addition, vision loss is linked with a "macular depression sign," representing an atrophic thinning macula. Yet, the most common cause for vision loss is associated with the proliferative stages of SCR.
Proliferative SCR is divided into five stages:8-10 Stage I is associated with microvascular occlusions at the level of the arteriole-capillaries. Arteriovenous anastomoses that develop to compensate for retinal non-perfusion characterizes stage II. Arteriovenous anastomoses are shunts, bridging areas of non-perfused retina and perfused retina within the peripheral retina (see figure 1).
Retinal non-perfusion leads to the development of stage III proliferative SCR, which is associated with peripheral retinal neovascularization. A retinal sea fans configuration is the classic feature of stage III proliferative SCR (see figure 2). Progression of the disease is often associated with neovascularization, which involutes, leading to fibrovascular tissue formation. (See "Sickle-Cell Disease Suspect," below.)
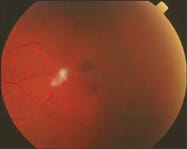
Fig 2: fibrovascularization associated with proliferative SCR.
Complications of neovascularization, leading to severe vision loss: vitreal hemorrhages (stage IV) and tractional retinal detachment (stage V) (see figure 3.)
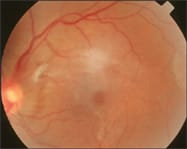
Fig 3: macular pseudohole associated with tractional retinal detachment.
Of note: "Dark without pressure" is a common benign retinal sign associated with sickle cell disease. If you note "dark without pressure" in a black or Caribbean patient who's not diagnosed with sickle cell disease, you should refer the patient to his primary-care physician for further evaluation.
| Sickle Cell Disease Suspect |
|---|
| A 29-year-old black woman presented complaining of decreased distance vision O.U. Her medical history was remarkable for sickle cell trait. Past ocular history was unremarkable. Exam Findings The patient's BCVA was 20/20 O.D. and O.S. with a small myopic prescription. Her slit lamp exam was unremarkable. Both pupils were equal, round and reactive to light with no afferent papillary defect. Dilated retinal evaluation, however, revealed an area of involuted neovascularization associated with fibrovascular tissue in the temporal retina O.D. (see figure 4), active neovascularization in the peripheral superior temporal retina (see figure 2) and arteriovenous anastomoses O.S. 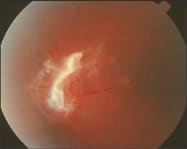
Fig 4: Proliferative SCR associated with peripheral sea-fan neovascularization. Notice the area of fibrovascularization. Diagnosis As a result of our findings, we diagnosed this patient with Stage III proliferative SCR. Management We referred the patient to her primary-care physician for continuous management and further re-evaluation for sickle cell disease. Also, we recommended a consult with a retina specialist. Unfortunately, we lost this patient to follow-up. |
Management
If you suspect sickle cell disease in a patient, obtain a thorough medical history of the individual, including genetic testing for the patient and his relatives. Also, refer the patient to his primary-care physician for screening and laboratory tests, such as a complete blood count (CBC), sickledex, peripheral blood smear and plasma hemoglobin electrophoresis.2,3
The ocular management of SCR depends on the type. Of note: Use carbonic anhydrase inhibitors (CAI) with caution in all sickle cell disease patients who have glaucoma, as these drugs have the potential to exacerbate the sickling process.
If the patient presents with non-proliferative SCR, schedule him for routine follow-up visits, typically between six to 12 months.
If the patient presents with proliferative SCR, fluorescein angiography helps determine the extent of capillary non-perfusion and associated neovascularization and may help guide appropriate treatment (explained below). Early intervention is critical for a good prognosis. (See "Sickle Cell Anemia Patient," above.)
For years, the conventional treatment of proliferative SCR was laser photocoagulation.11-12 Surgical options, such as scleral buckling and pars plana vitrectomy, are reserved for retinal detachments and long-standing vitreous hemorrhage, respectively.
Although neovascularization associated with proliferative SCR has shown spontaneous regression in 67% of cases, studies have reported that 12% of untreated cases result in a decrease of central vision.13
More recent studies have examined the role of vascular endothelial growth factors (VEGF) in the development of neovascularization associated with proliferative SCR. One study revealed the presence of VEGF in the development of neovascularization associated with proliferative SCR.14
Another study showed that neovascularization regressed after the use of intravitreal bevacizumab (Avastin, Genentech), a potent anti-VEGF therapy.15 Furthermore, a study reported a case of neovascularization and a vitreal hemorrhage resolving following the intravitreal injection of bevacizumab.16
Because the ocular complications of sickle cell disease may cause significant vision loss and be the initial indication of the disease itself, we, as optometrists, play an important role in the management of these patients. Remember: The prognosis of both sickle cell disease and SCR is favorable with early recognition and prompt treatment, when indicated. OM
1. Sickle Cell Disease Association of America, Inc. Sickle Cell Facts. How many people have sickle cell disease? www.sicklecelldisease.org/about_scd/faqs.phtml#people (Accessed Nov.12, 2008)
2. J.B. Schnog, A.J. Duits and F.A.J. Muskiet et al., Sickle cell disease; a general overview, Neth J Med. 2004 Nov;62(10): 364–74. Review.
3. Sickle Cell Disease Guideline Panel. Sickle cell disease: screening, diagnosis, management, and counseling in newborns and infants. Clinical Practice Guideline no. 6. Rockville, MD: Agency for Health Care Policy and Research, U.S. Public Health Service, 1993. AHCPR publication no. 93-0562.
4. Ashley-Koch A, Yang Q, Oiney RS. Sickle hemoglobin (Hb S) allele and sickle cell disease: a HuGE review. Am J Epidemiol 2000 May 1;151(9):839-45. Review.
5. Reynolds SA, Besada E, Winter-Corella C. Retinopathy in patients with sickle cell trait. Optometry. 2007 Nov;78(11):582-7.
6. Paton D. The conjunctival sign of sickle-cell disease. Arch Ophthalmol. 1961 Jul;66:90–94.
7. Serjeant G.R. The sickle cell trait. In: G.R. Serjeant, Editor, Sickle cell disease (ed 2), New York: Oxford University Press, 1992: pp. 415–425.
8. Clarkson J.G. The ocular manifestations of sickle-cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc. 1992;(90):481–504.
9. Goldberg MF. Classifications and pathogenesis of proliferative sickle retinopathy. Am J ophthalmol. 1971 Mar;71(3):649-65.
10. Cohen SB, Fletcher ME, Goldberg MF, Jednock NJ. Diagnosis and management of ocular complications of sickle hemoglobinopathies — Part I, Ophthalmic Surg. 1986 Jan;17(1):57–59.
11. Rednam KR, Jampol LM, Goldberg MF. Scatter laser photocoagulation for proliferative sickle cell retinopathy. Am J Opthalmol. 1982; May93(5):594-9.
12. Jacobson MS, Gagliano DA, Cohen SB, et al. A randomized clinical trial of feeder vessel photocoagulation of sickle cell retinopathy. Opthalmol. 1991 May;98(5):581-5.
13. Codon PI, Serjeant GR. Behavior of untreated PSCR. Br J Ophthalmol. 1980; 64: 404-11.
14. Cao J, Mathews MK, McLeod DS, et al. Angionenic factors in human proliferative sickle cell retinopathy, Br J Ophthalmol. 1999 Jul;83(7):838-46.
15. Siqueira C, Costa RA, Scott IU, et al. Intravitreal bevacizumab (Avastin) injection associated with regression of retinal neovascularization caused by sickle cell retinopathy. Acta Ophthalmol Scand. 2006 Dec;84(6):834-5.
16. Shaikh S. Intravitreal bevacizumab (Avastin) for the treatment of proliferative sickle retinopathy. Indian J Ophthalmol. 2008 May-Jun;56(3):259.

|
Dr. Espejo is an assistant professor of optometry at Nova Southeastern University College of Optometry. She currently serves as an attending physician at the eye institute and as the director of Externships. E-mail her at aespejo@nova.edu. |

|
Dr. Shechtman is an associate professor at Nova Southeastern University College of Optometry in Ft. Lauderdale and works at the eye institute and diabetic/macula clinic. She is a member of the Optometric Retinal Society (ORS). E-mail her at dianashe@nova.edu. |

|
Dr. Reynolds is an assistant professor at Nova Southeastern University and serves as a module director and attending optometric physician at the eye institute and diabetic/macula clinic. She has authored numerous ocular disease posters and has lectured on a local and national level. E-mail her at sreynold@nova.edu. |