


Because inherited retinal disease (IRD) has historically been a complex and challenging area, IRD suspects have been referred to teaching hospitals and research settings, which is not a feasible journey for many of them. Thanks to recent advances in multimodal imaging, functional testing, genetic testing, and management strategies, however, IRD care is now accessible to the conveniently located optometrist.
Multimodal Imaging
Widefield color photography, fundus autofluorescence (FAF), optical coherence tomography (OCT), near-infrared imaging (NIR) on newer OCT models, fluorescein angiography, and indocyanine green angiography are beneficial in identifying IRDs. Widefield imaging ensures a detailed evaluation of both the central and peripheral retina. FAF provides insights into retinal pigment epithelium (RPE) health and function.
NIR uses long wavelengths of light that penetrate deep into the retina to improve visualization of sub-retinal and choroidal abnormalities in sometimes surprising detail. Finally, fluorescein angiography and indocyanine green angiography can be used for identifying abnormal neovascularization. These are less commonly used now due to the advent of OCT angiography (OCTA).
A 60-year-old-male recently presented to my practice as a new patient with a history of advanced dry age-related macular degeneration (AMD). He said he was trying a different practice because he was frustrated with his vision. I received fundus photography and OCT images, as well as notes from the patient’s prior eyecare provider.
At the patient’s visit with me, he said he had no family history of vision loss that he was aware of, and that he was compliant with an ocular nutritional supplement because of the dry AMD diagnosis.
When examining his retinas, I noted that his diagnosis was very advanced and unusual for his age. This was my first suspicion that it could be an AMD masquerader. As a result, I ordered additional testing.
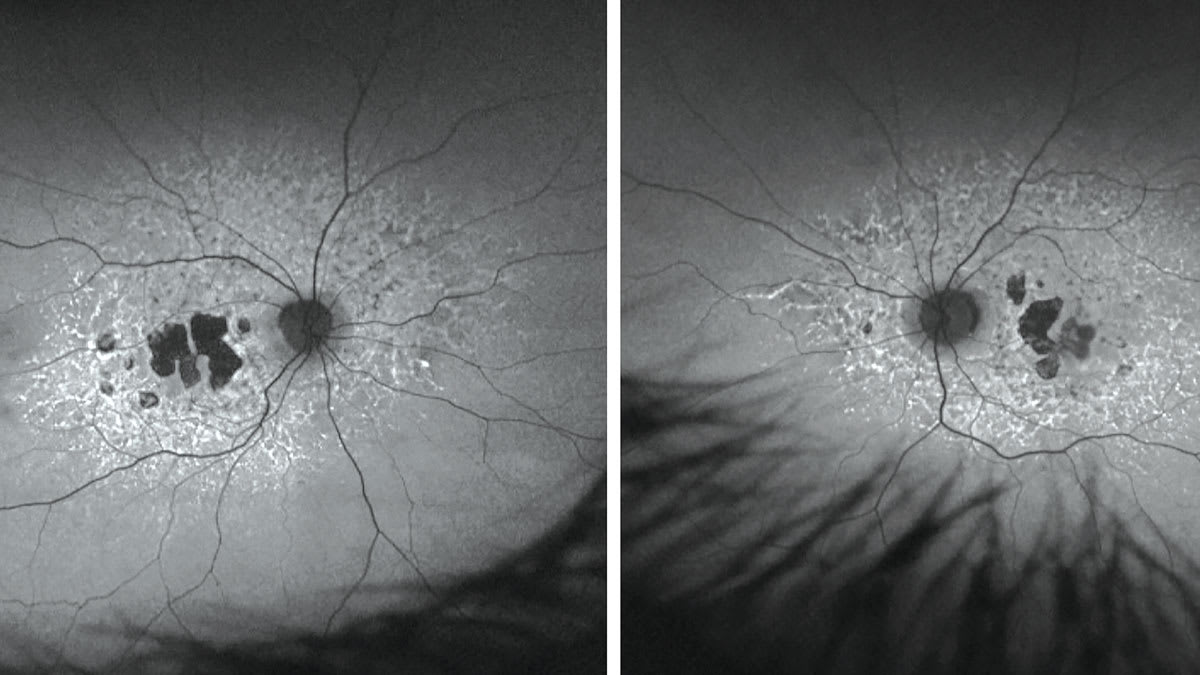
FAF imaging was inconsistent with AMD (Figure 1). Although AMD shows RPE disturbances and atrophy, these findings are typically confined to the macula. This FAF showed patchy areas of central atrophy (hypoFAF), which could fit with AMD. The abnormality that set these images apart was the symmetric hyperFAF branches and flecks that were paramacular and extending beyond the vascular arcade of the posterior pole. The symmetrical and unusual hyperFAF pattern led me to believe the patient likely had an IRD.
OCT provided a structured analysis of individual retina layers (OCTA is helpful in identifying early choroidal neovascularization [CNV]). Notably, Many IRDs carry an increased risk for CNV, so proactively diagnosing this early and initiating anti-vascular endothelial growth factor treatment provides better long-term visual outcomes.
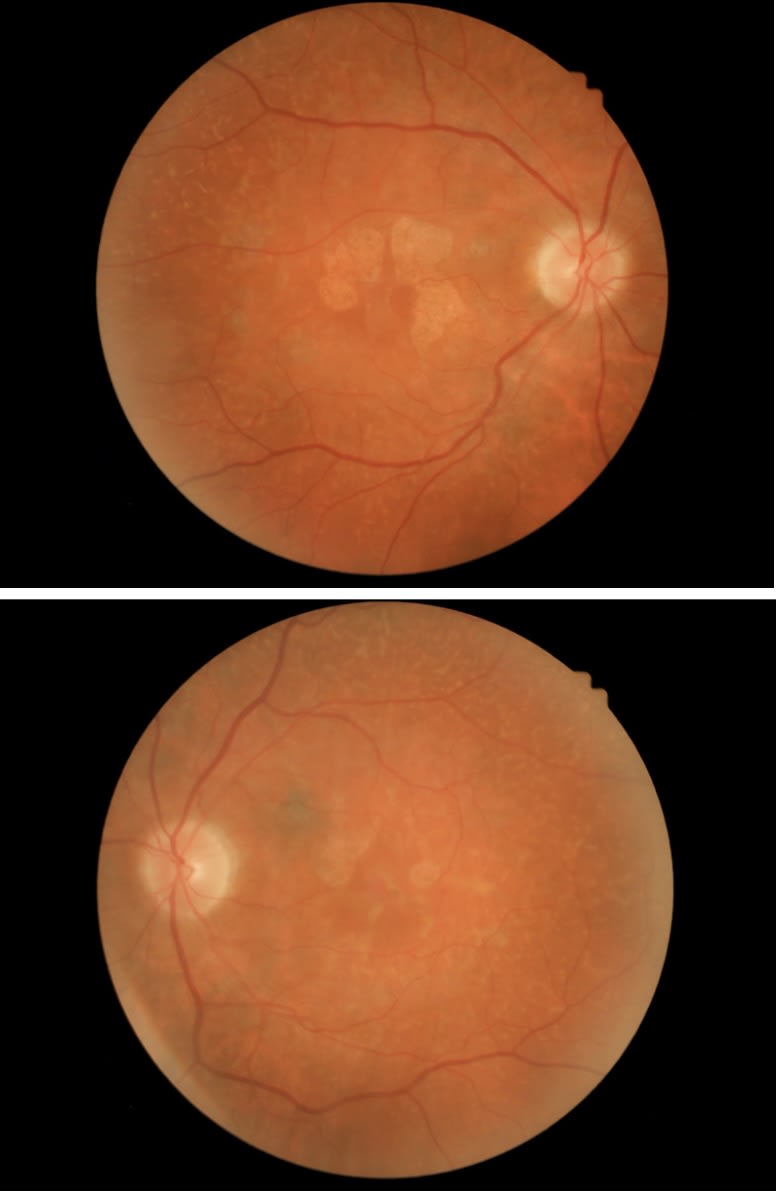
This patient’s OCT showed large areas of atrophy (see images below), which matches the areas of hypoFAF. It also revealed deposition anterior to the RPE that is unlike drusen—another clue that this was an AMD masquerader.
Functional Testing
Full-field electroretinography (FFERG) is beneficial in identifying IRDs, as it aids in evaluating retinal function objectively and can detect dysfunction before visual symptoms become apparent. Specifically, since AMD mainly affects the central retina, it is unlikely to affect FFERG values. However, a more global retinal disease does cause abnormalities in FFERG testing. This is especially true with retinitis pigmentosa patients. I did not employ FFERG on the mentioned patient, as I saw him prior to my practice acquiring this technology.
Dark adaptation (DA) is severely delayed in many IRDs, such as retinitis pigmentosa. Visual field (VF) testing is helpful for monitoring disease progression, and the pattern of VF deficits can provide details of a patient’s functional status: Are there peripheral field constriction and/or central VF deficits, and how are they changing over time?
Because advancing AMD is also known to affect DA, I did not use dark adaptometry on this patient. That said, DA is useful in early IRD diagnoses. For example, a young person who has peripheral pigmentary changes should still have a normal DA if the pigmentary changes are benign. However, if this is an early sign of retinitis pigmentosa, the DA will be significantly abnormal and won’t match another diagnostic cause.
The other functional tests of color vision testing and contrast sensitivity can provide a patient’s full clinical picture, which is always helpful. As a reminder: Visual acuity (VA) does not provide information on the patient’s total visual experience.
This patient had small islands of a healthy central retina, so his VA was 20/50 OD and 20/30 OS at the time of this imaging. He said his overall functioning was much less in reading, using the computer, checking his phone, and in driving, due to glare. I recommended genetic testing, which the patient underwent.
Genetic Testing
Results showed a pathogenic variant in the patient’s PRPH2 gene, which can cause a host of IRDs, including pattern dystrophies, reticular dystrophy, central areolar choroidal dystrophy, and cone and cone-rod dystrophies.1
Review of the patient’s genotype and phenotype showed the patient had pseudo-Stargardt pattern dystrophy (PSPD) and not dry AMD.
PSPD resembles Stargardt disease but differs in several key ways: Onset typically occurs later in life (commonly in the 40s to 50s rather than in childhood), progression is slower and more variable, and inheritance is often autosomal dominant, whereas Stargardt disease is more often autosomal recessive.
On fundus autofluorescence (FAF), both IRDs may show fleck-like patterns, but PSPD tends to demonstrate scattered flecks that have variable hyperFAF intensity, in contrast to the more uniform, well-defined pisciform pattern of Stargardt.

Although PSPD is inherited in an autosomal dominant manner, this patient had no reported family history of vision loss. Importantly, PRPH2-associated retinal conditions are known for their wide variability—even with the same mutation, and even within the same family. Thus, individuals may differ significantly in symptoms, age of onset, and disease course, with some remaining asymptomatic throughout life. This variability is also characteristic of other IRDs, such as Best disease. Thus, the absence of a known family history does not exclude the possibility of an IRD.
Today’s genetic testing can confirm the genotype and inheritance patterns of the IRD, facilitates more accurate prognosis about disease progression, and, perhaps most importantly, identifies eligibility for clinical trials and emerging therapies.
Genetic research, particularly in IRDs, is rapidly advancing. Just over the last decade, more than 100 new IRD-related genes have been identified. To date, approximately 300-known genes play roles in various IRDs, and current statistics show roughly 70% of IRDs diagnosed in a clinical setting can now be accurately genotyped through genetic testing.2
Modern IRD testing panels use next-generation sequencing to detect point mutations, insertions/deletions, and some copy number variants across hundreds of IRD-associated genes3. Genetic testing that clearly identifies the genetic cause of disease does not need to be repeated, but testing that was negative or inconclusive upon original testing should be revisited in the future, as research and technology continue to evolve.
If genetic testing was performed more than 5 years ago, patients may benefit from repeat testing with updated gene panels and improved analytics.4 The American Academy of Ophthalmology Task Force for Genetic Testing recommends that all patients with suspected inherited eye disease undergo high-quality genetic testing that is interpreted alongside medical geneticists and genetic counselors.5
Any US eye doctor can be a provider of free genetic testing through the My Retina Tracker website, which is sponsored through Foundation Fighting Blindness (see sidebar, “Eligible Inherited Retinal Disease Diagnoses”). Specifically, a saliva or buccal sample is taken at the office, patients sign paperwork electronically through DocuSign, and the sample is mailed to the testing site. Results take 4 to 6 weeks.
Through this process, patients are also signed up for the My Retina Tracker Registry, which provides updates on relevant research studies or clinical trials they may be eligible for based on their Registry profile.
Once the results are ready, patients can schedule their free session with a genetic counselor to discuss their results. A detailed report is then given to the ordering doctor that includes the results of the testing and the counseling.
Management Strategies
Optimizing visual function—ocular health management, a low vision evaluation, lens tints, using prism or task-specific glasses—is the optometric management strategy for IRD patients. To arrive at these interventions, we must first ask the patient how the IRD affects their daily function and mental health.
The 60-year-old patient was referred to an optometrist who provides low-vision services. This OD prescribed near-vision-only glasses in place of the patient’s current progressive lenses. Additional recommendations included the use of proper task lighting, education on technology-based tools (such as phone accessibility features and downloadable applications), and tinted lenses for both indoor and outdoor use to address the glare affecting his driving.
Because the patient had expressed significant awareness of his visual decline over the past year and reported anxiety regarding the potential for further vision loss or complete blindness, I referred him to our local Vision Resource Center. This Center provides low vision–focused mental health counseling. He has also participated in group therapy with peers experiencing vision loss at a similar age. He says it has provided him with both reassurance and support. Incidentally, we should consider providing a list of social media groups for IRD patients to connect with others experiencing the same diagnosis and challenges. I have patients with IRDs who have found various online support groups and find comfort knowing they are not alone.
FightingBlindness.org provides updates on IRD scientific research, vision loss support, and educational opportunities, such as webinars and podcasts.
IRD patients may also benefit from having a guide dog or be eligible for state services if they have low vision or an eye disease expected to progress. (See https://www.guidedogs.com/). Also, VisionAware.org provides low vision resource options throughout the United States. Another website of value: Retina International (https://retina-internation al.org).
Scheduling follow-up appointments with IRD patients is important for monitoring disease progression and discussing evolving difficulties with daily-living activities. It’s worth mentioning to these patients that gene therapy may be the future of IRD management. Specifically, the eye is easily accessible for injections and surgical interventions and has immune-privileged status, facilitating viral vector accommodation. Tight blood-ocular barriers prevent systemic contamination, making the eye an ideal target organ.
In addition, because retinal tissue consists of neural cells that do not regenerate or turn over, successful gene therapy would theoretically provide long-term benefits without requiring repeated treatments—unlike other organs where cell turnover necessitates ongoing therapy.6 As a result, we should both keep tabs on and recommend that patients and their families check out www.clinicaltrials.gov.
Luxturna (voretigene neparvovec, Spark Therapeutics) is a gene therapy for biallelic RPE65 mutations and is currently in clinical trials for targeting other common genes affected in IRDs, including RPGR, PRPH2, and USH2A4.
Let's Dig In
Optometrists’ scope perfectly aligns with IRD management needs: diagnosing and managing eye disease from early detection while offering comprehensive visual rehabilitation strategies, patient education, advocacy, and support through the patient’s journey. OM
References
- Conley SM, Naash M. Gene therapy for PRPH2-associated ocular disease: challenges and prospects. Cold Spring Harb Perspect Med. 2014;4(11):a017376. doi: 10.1101/cshperspect.a017376.
- Georgiou, M et al. Phenotyping and genotyping inherited retinal diseases: molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, Leber congenital amaurosis, and cone dysfunction syndromes. Prog Retin Eye Res. 2024 May: 100:101244. DOI: 10.1016/j.preteyeres.2024.101244
- Schneider, N et al. Inherited retinal diseases: linking genes, disease-causing variants, and relevant therapeutic modalities. Prog Retin Eye Res. 2022 Jul: 80:101029. DOI: 10.1016/j.preteyeres.2021.101029.
- Goetz, K et al. Genetic testing for inherited eye conditions in over 6,000 individuals through the eyeGENE network. Am J Med Genet C Semin Med Genet. 2020; 184(3): 828-837. DOI: 10.1002/ajmg.c.31843.
- Stone, EM et al. Recommendations for genetic testing of inherited eye diseases: report of the American Academy of Ophthalmology Task Force on Genetic Testing. Ophthalmology. 2012; 119:2408-2410.
- Dockery, A et al. Next-generation sequencing applications for inherited retina disease. Int J Mol Sci. 2021 May 26;22(11):5684. DOI: 10.3390/ijms22115684.
